Nội dung bài
NHẮC LẠI CHỐNG CHỈ ĐỊNH SỬ DỤNG THUỐC ỨC CHẾ MEN CHUYỂN (ACEI) VÀ THUỐC ỨC CHẾ THỤ THỂ ANGIOTENSIN (ARB) TRONG THAI KỲ: THÔNG TIN TỪ CƠ QUAN AN TOÀN THUỐC VÀ THIẾT BỊ Y TẾ NEW ZEALAND (MEDSAFE)
Trung tâm theo dõi phản ứng có hại ở New Zealand (CARM) đã nhận được một báo cáo nghi ngờ việc sử dụng đồng thời losartan và empagliflozin trong thai kỳ dẫn đến phản ứng bất lợi trên thai nhi. Các phản ứng được báo cáo gồm hội chứng suy thai, rối loạn thai nhi và hội chứng Potter (một tình trạng hiếm gặp làm giảm nước ối và suy thận ở thai nhi). Dữ liệu về tính an toàn của empagliflozin trong thai kỳ còn hạn chế. Điểm tin này nhắc lại các thuốc ACEi/ARB bị chống chỉ định trong thai kỳ.
Tăng huyết áp khi mang thai
Trong thai kỳ, huyết áp không được kiểm soát có thể dẫn đến tiền sản giật và những hậu quả bất lợi cho cả mẹ và thai nhi.
Thuốc điều trị tăng huyết áp được khuyến cáo sử dụng cho phụ nữ mang thai gặp cơn tăng huyết áp nghiêm trọng nhằm giảm huyết áp nhanh chóng. Thuốc cũng được cân nhắc sử dụng trên phụ nữ có tăng huyết áp thai kỳ, đặc biệt khi có thêm các yếu tố nguy cơ gây tiền sản giật và/hoặc các bệnh đi kèm.
Nguy cơ khi sử dụng ACEi/ ARB
Các thuốc đầu tay trong điều trị tăng huyết áp ở bệnh nhân người lớn bao gồm ACEi (ví dụ: enalapril, lisinopril, perindopril, quinepril, ramipril) và ARB (ví dụ: candesartan, losartan, telmisartan). Tuy nhiên, các thuốc này bị chống chỉ định trong thai kỳ. Việc sử dụng các thuốc này trong thời kỳ mang thai có liên quan đến độc tính trên thai nhi và trẻ sơ sinh, bao gồm dị tật hộp sọ, giảm thể tích dịch ối, hạ huyết áp, tăng kali máu, suy thận và thai chết lưu.
Khuyến cáo khi kê đơn ACEi/ ARB cho bệnh nhân trong độ tuổi sinh sản
- Trước khi bắt đầu điều trị bằng các thuốc ACEi/ ARB, bác sĩ cần hỏi bệnh nhân có đang mang thai hoặc có dự định mang thai hay không.
- Thông báo cho bệnh nhân về nguy cơ của các thuốc ACEi/ ARB lên thai nhi và nhắc bệnh nhân đến gặp bác sĩ ngay khi có thai. Nếu bệnh nhân có kế hoạch mang thai, cân nhắc chuyển sang sử dụng thuốc điều tăng huyết áp khác thay thế trước khi thụ thai.
- Nếu bệnh nhân mang thai trong quá trình điều trị bằng các thuốc ACEi/ ARB, cần ngừng thuốc và thay thế bằng thuốc điều tăng huyết áp khác phù hợp.
---------------------------------------------------------------------------------------------------------------------------------------------------------------------
HỘI CHỨNG VIÊM RUỘT LIÊN QUAN ĐẾN KHÁNG SINH AMOXICILLIN: THÔNG TIN TỪ BỘ Y TẾ CANADA (HEALTH CANADA)
Hội chứng viêm ruột do thuốc là một phản ứng dị ứng, với triệu chứng điển hình là nôn kéo dài (xuất hiện trong vòng 1-4 giờ sau khi dùng thuốc), chủ yếu được báo cáo ở trẻ em sử dụng các thuốc chứa amoxicillin. Các triệu chứng khác có thể bao gồm đau bụng, mệt mỏi, tiêu chảy, hạ huyết áp hoặc tăng bạch cầu trung tính. Trong các trường hợp nghiêm trọng, hội chứng viêm ruột do thuốc có thể tiến triển thành sốc.
Bệnh nhân được khuyến cáo ngừng sử dụng thuốc chứa amoxicillin và thông báo ngay cho nhân viên y tế nếu gặp các triệu chứng của hội chứng viêm ruột liên quan đến thuốc do đây có thể là dấu hiệu của phản ứng dị ứng nghiêm trọng.
Nguy cơ gặp hội chứng viêm ruột sẽ được cập nhật trong các tờ thông tin sản phẩm của thuốc chứa amoxicillin tại Canada. Thông tin này sẽ được cập nhật vào các mục Cảnh báo và thận trọng, Tác dụng không mong muốn trong thông tin sản phẩm của các thuốc chứa amoxicillin lưu hành tại Canada.
---------------------------------------------------------------------------------------------------------------------------------------------------------------------
BỆNH PHỔI KẼ DO THUỐC: THÔNG TIN TỪ MEDSAFE
Tại cuộc họp vào tháng 09/2024, Hội đồng đánh giá phản ứng có hại của thuốc (Medicines Adverse Reactions Committee – MARC) của Cơ quan An toàn Thuốc và Thiết bị Y tế New Zealand (Medsafe) đã đánh giá một báo cáo ca viêm phổi không đặc hiệu liên quan đến methotrexat. Trong điểm tin này, Medsafe nhắc lại về nguy cơ mắc bệnh phổi kẽ khi sử dụng methotrexat và các thuốc khác.
Thuốc gây bệnh phổi kẽ
Bệnh phổi kẽ là thuật ngữ chỉ một nhóm bệnh lý gây viêm hoặc xơ hóa nhu mô phổi. Những tổn thương này làm suy giảm khả năng trao đổi khí của phổi và có thể dẫn đến suy hô hấp hoặc tử vong.
Bệnh phổi kẽ là phản ứng có hại của thuốc trên phổi phổ biến nhất. Một số thuốc có thể gây bệnh phổi kẽ gồm nitrofurantoin, methotrexat, amiodaron, leflunomid, hóa trị liệu và một số thuốc sinh học (Bảng 1). Medsafe đã ghi nhận báo cáo của hàng trăm thuốc có khả năng gây bệnh phổi kẽ.
Bảng 1. Ví dụ về các thuốc liên quan đến bệnh phổi kẽ
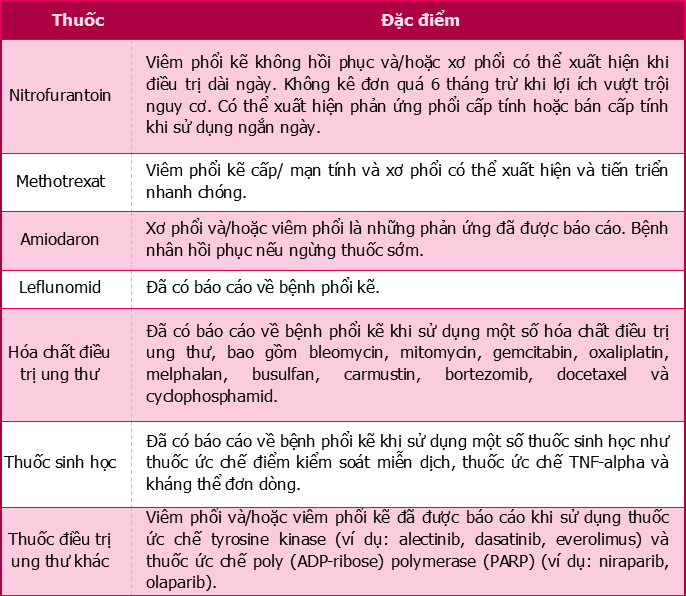
Giám sát bệnh phổi kẽ do thuốc
Các triệu chứng khởi phát của bệnh phổi kẽ không đặc hiệu, bao gồm ho, khó thở và mệt mỏi. Chẩn đoán bệnh phổi kẽ dựa trên các dấu hiệu lâm sàng, hình ảnh học và mô học. Trên lâm sàng, thuốc có thể là một nguyên nhân gây bệnh phổi kẽ khi bệnh nhân xuất hiện các triệu chứng hô hấp mới hoặc tiến triển trầm trọng hơn trong thời gian dùng thuốc.
Yếu tố nguy cơ gây bệnh phổi kẽ do thuốc:
- Tuổi (trẻ em và người cao tuổi có nguy cơ gặp phản ứng có hại cao hơn).
- Bệnh lý nền liên quan đến phổi.
- Tương tác thuốc: sử dụng đồng thời các thuốc có thể gây bệnh phổi kẽ.
Xử trí viêm phổi kẽ
Khi sử dụng các thuốc có nguy cơ gây bệnh phổi kẽ, cần theo dõi chặt chẽ chức năng hô hấp của bệnh nhân. Phát hiện bệnh phổi kẽ muộn làm tăng nguy cơ tổn thương phổi không hồi phục, thậm chí tử vong. Nhân viên y tế cần tham khảo hướng dẫn sử dụng thuốc và các hướng dẫn điều trị liên quan để có biện pháp xử trí phù hợp.
Khi phát hiện bệnh nhân mắc bệnh phổi kẽ do thuốc, cần ngừng thuốc ngay và điều trị bằng corticosteroid. Việc nhận biết và điều trị sớm có thể cải thiện tiên lượng của bệnh nhân.
Tư vấn cho bệnh nhân về nguy cơ bệnh phổi kẽ: Bệnh nhân sử dụng các thuốc có thể gây bệnh phổi kẽ cần được thông báo về nguy cơ này. Khuyến cáo bệnh nhân đến cơ sở y tế ngay khi xuất hiện các triệu chứng như ho, đau ngực, khó thở, sốt hoặc ớn lạnh. Giải thích cho bệnh nhân rằng việc phát hiện và điều trị sớm rất quan trọng để giảm nguy cơ mắc tổn thương phổi không hồi phục.
---------------------------------------------------------------------------------------------------------------------------------------------------------------------
NGUY CƠ HẠ PHOSPHAT MÁU KHI SỬ DỤNG SẮT ĐƯỜNG TĨNH MẠCH: THÔNG TIN TỪ MEDSAFE
Nguy cơ hạ phosphat máu
Hạ phosphat máu là một phản ứng có hại đã biết khi sử dụng sắt đường tĩnh mạch, đặc biệt là sắt carboxymaltose. Hạ phosphat máu có thể diễn biến nghiêm trọng, kéo dài và có thể gây ra các biến chứng như loãng xương và gãy xương. Cần đánh giá nguy cơ hạ phosphat máu ở bệnh nhân trước khi chỉ định sắt đường tĩnh mạch.
Sắt đường tĩnh mạch được chỉ định trong điều trị và/hoặc dự phòng thiếu sắt khi các chế phẩm sắt đường uống không phù hợp hoặc không hiệu quả. Các chế phẩm sắt đường đường tĩnh mạch đang được cấp phép lưu hành tại New Zealand bao gồm sắt carboxymaltose và sắt polymaltose. Hiện nay, việc sử dụng sắt đường tĩnh mạch, đặc biệt là sắt carboxymaltose đang ngày càng gia tăng ở New Zealand. Do đó, Medsafe đã đưa cảnh báo về nguy cơ hạ phosphat máu sau khi sử dụng sắt đường tĩnh mạch.
Cơ chế hạ phosphat máu
Cơ chế của hạ phosphat máu khi sử dụng sắt đường tĩnh mạch do sự gia tăng yếu tố tăng trưởng nguyên bào sợi (FGF23), dẫn đến thận bài tiết phosphat quá mức và làm giảm nồng độ phosphat máu.
Mức độ nghiêm trọng
Tình trạng hạ phosphat máu do sử dụng sắt đường tĩnh mạch thường thoáng qua và không có triệu chứng. Tuy nhiên, hạ phosphat máu nghiêm trọng, kéo dài, đồng thời xuất hiện các biến chứng như loãng xương và gãy xương cũng có thể xảy ra, đặc biệt ở những bệnh nhân có các yếu tố nguy cơ.
Yếu tố nguy cơ gây hạ phosphat máu
Các yếu tố nguy cơ gây hạ phosphat máu liên quan đến chế phẩm chứa sắt đường tĩnh mạch được trình bày ở Bảng 2. Bệnh nhân cần sử dụng sắt đường tĩnh mạch dài hạn có nguy cơ cao hạ phosphat máu và gặp các biến chứng của hạ phosphat máu.
Bảng 2. Các yếu tố nguy cơ gây hạ phosphat máu khi sử dụng sắt đường tĩnh mạch

Cân nhắc khi kê đơn
Khi kê đơn sắt đường tĩnh mạch cần:
- Lưu ý thông tin về tác dụng không mong muốn của các chế phẩm sắt khác nhau và các yếu tố nguy cơ hạ phosphat máu trên bệnh nhân.
- Thông báo cho bệnh nhân về nguy cơ hạ phosphat máu và các biểu hiện thường gặp như đau xương, đau khớp, mệt mỏi.
- Theo dõi nồng độ phosphat ở những bệnh nhân có nguy cơ cao gặp hạ phosphat máu hoặc các biến chứng liên quan.
- Đánh giá lại việc điều trị bằng sắt đường tĩnh mạch khi xuất hiện biến cố hạ phosphat máu.
---------------------------------------------------------------------------------------------------------------------------------------------------------------------
THÔNG TIN CẬP NHẬT VỀ THUỐC OCALIVA (ACID OBETICHOLIC) TỪ CƠ QUAN QUẢN LÝ DƯỢC PHẨM CHÂU ÂU (EMA) VÀ CƠ QUAN QUẢN LÝ DƯỢC PHẨM VÀ THỰC PHẨM HOA KỲ (US.FDA)
Cơ quan Quản lý Thực phẩm và Dược phẩm Hoa Kỳ (US.FDA)
Ngày 12/12/2024, US.FDA đưa ra cảnh báo cho nhân viên y tế về nguy cơ tổn thương gan nghiêm trọng khi sử dụng Ocaliva (acid obeticholic) để điều trị viêm đường mật nguyên phát trên bệnh nhân không mắc xơ gan. Dựa trên đánh giá các dữ liệu thử nghiệm lâm sàng hậu mãi, US.FDA đã phát hiện các ca tổn thương gan nghiêm trọng trên bệnh nhân không mắc xơ gan, dẫn đến cần phải ghép gan. Bệnh nhân cần được theo dõi chức năng gan định kì để phát hiện sớm tình trạng suy giảm chức năng gan và ngừng thuốc Ocaliva kịp thời.
Ocaliva (acid obeticholic) là thuốc kê đơn đã được US.FDA phê duyệt vào tháng 05/2016. Trong thử nghiệm lâm sàng, thuốc cho thấy tác dụng làm giảm alkaline phosphatase (ALP) trên bệnh nhân viêm đường mật nguyên phát không đáp ứng tốt với acid ursodeoxycholic.
Năm 2021, FDA đã giới hạn chỉ định của Ocaliva trên bệnh nhân viêm đường mật nguyên phát có xơ gan tiến triển do nguy cơ tổn thương gan nghiêm trọng trên đối tượng này. Mục Chống chỉ định trong thông tin sản phẩm của thuốc này đã được cập nhật. Tuy nhiên, một số báo cáo ca gần đây gửi tới FDA vẫn có trường hợp bệnh nhân sử dụng Ocaliva kể cả khi có chống chỉ định.
Sau khi thuốc được lưu hành trên thị trường, US.FDA đã yêu cầu tiến hành thêm các thử nghiệm lâm sàng hậu mãi để đánh giá lợi ích trên lâm sàng của Ocaliva. Trên nhóm đối tượng bệnh nhân sử dụng thuốc theo chỉ định được phê duyệt, nguy cơ ghép gan và tử vong khi sử dụng Ocaliva cao hơn so với sử dụng giả dược. Đặc biệt, trong nhóm bệnh nhân được chỉ định Ocaliva là nhóm có nguy cơ tiến triển ghép gan hoặc tử vong thấp hơn, 07/81 bệnh nhân sử dụng Ocaliva cần phải ghép gan, so với 01/68 bệnh nhân sử dụng giả dược (trường hợp bệnh nhân ghép gan trong nhóm giả dược đã sử dụng Ocaliva 2 năm trước khi ghép gan, do vậy không loại trừ mối liên quan với thuốc). 04 bệnh nhân nhóm sử dụng Ocaliva tử vong, so với 01 bệnh nhân nhóm giả dược. Tỷ suất nguy cơ (hazard ratio - HR) ghép gan và tử vong trên bệnh nhân không có xơ gan tiến triển và không thuộc chống chỉ định là 4,77 (95% CI 1,03-22,09).
Sau khi mở rộng chống chỉ định của thuốc trên bệnh nhân viêm đường mật nguyên phát có xơ gan tiến triển, trong giai đoạn từ 26/05/2021 đến 18/09/2024, US.FDA đã ghi nhận 20 báo cáo ca (13 ca trong nước, 07 ca trên thế giới) gặp biến cố khi sử dụng Ocaliva: ghép gan (07 trường hợp), đang đánh giá hoặc chờ ghép gan (08 trường hợp) và tử vong do bệnh gan (06 trường hợp). Mặc dù thông tin từ báo cáo ca chưa đầy đủ để đánh giá việc sử dụng Ocaliva, US.FDA đã ghi nhận 03 trường hợp trong nước gặp biến cố bất lợi trên gan do tiếp tục sử dụng Ocaliva mặc dù xuất hiện bệnh gan tiến triển. Điều này cho thấy tầm quan trọng của việc theo dõi chức năng gan và ngừng thuốc nếu bệnh nhân xuất hiện dấu hiệu xơ gan tiến triển.
Nhân viên y tế cần theo dõi chức năng gan định kỳ trên bệnh nhân sử dụng Ocaliva nhằm phát hiện sớm tình trạng suy giảm chức năng gan. Cần ngừng sử dụng thuốc khi bệnh nhân có dấu hiệu của bệnh gan tiến triển hoặc thuốc không có hiệu quả. Bệnh nhân cần liên hệ với nhân viên y tế ngay khi xuất hiện các triệu chứng tổn thương gan.
Cơ quan Quản lý Dược phẩm châu Âu (EMA)
Trước đó, ngày 27/06/2024, Ủy ban sử dụng thuốc cho người (CHMP) của EMA đã quyết định rút giấy phép lưu hành của thuốc Ocaliva (acid obeticholic) do lợi ích không còn vượt trội nguy cơ. CHMP đã đánh giá kết quả của thử nghiệm 747-302, một thử nghiệm lâm sàng ngẫu nhiên đánh giá hiệu quả và độ an toàn của Ocaliva trên bệnh nhân không đáp ứng hoặc không sử dụng được acid ursodeoxycholic (UDCA) để điều trị viêm đường mật nguyên phát. Kết quả cho thấy Ocaliva không có hiệu quả hơn so với giả dược. Đồng thời, dữ liệu đời thực và từ các nghiên cứu quan sát chưa khẳng định được hiệu quả của thuốc. Do vậy, Ủy ban kết luận lợi ích của việc sử dụng Ocaliva không còn vượt trội so với nguy cơ thuốc gây ra và quyết định rút giấy phép lưu hành của thuốc này. Quyết định có hiệu lực pháp lý ở tất cả các quốc gia là thành viên của EU từ ngày 03/08/2024.
